SMILES 标准化算法 - CANGEN
CommentSMILES 是化学信息学中最常见的分子形式,其因为可读性强、体积小且“表格友好”而广受应用。但由于同一个分子可以有多种不同的 SMILES 形式导致我们不能将它用作化合物的匹配与去重操作。于是将 SMILES 标准化(Canonicalise)的需求就产生,如果可以通过某个算法保证一个化合物只会有一种 SMILES 自然可以增加 SMILES 在化学信息学中的作用。其实不难想象,SMILES 之所以可以出现不同是因为我们可以随意修改原子的“出场顺序”以得到不同的 SMILES,比如苯甲酸,我们可以先写苯环再写羧基,也可以先写羧基再写苯环,即使先写苯环也可以考虑以不同的原子为起点,可以顺时针或者逆时针。所以如果我们能通过某个算法给这些原子排序,谁序号大则优先出场,SMILES 自然就可以保证唯一。
1 | 苯甲酸的几种写法 |
原子排序
我们可能马上可以想到 CIP 排序,即 IUPAC 用来判断手型时对原子进行的排序。这样排序可能不太好,比如乙醚,按照 CIP 排序则是氧最大,会写成O(CC)CC,明明可以线性描述CCOCC偏偏给氧增加“支链”。其次 CIP 排序需要遍历支链,效率上可能不太友好。这里我们介绍下比较经典的 CANGEN 算法,这个名字其实之前那篇 扩展连通性指纹(ECFP/Morgan Fingerprint)介绍 就有提到过,它给原子排序的原理其实和扩展连通性指纹类似,只是扩展连通性指纹只是为了保证“不同环境下的原子有唯一的标识符”,于是会用到哈希算法而且迭代次数是超参,而标准化 SMILES 则要求不同环境下原子有唯一的排序,因此需要迭代到收敛为止(即如果排序一样说明是对称结构)。
根据文献,该算法过程大致如下:1
2
3
4
5
6
7
8
9
10
11(1) 首先给每个原子初始化“识别符”,跳过第二步直接进入第三步
------- 以下过程循环 -------
(2) 用周边原子排序的“质数积”描述该原子,得到新向量
(3) 对该向量进行排序,但要维持上一次的排序(即只关心上次排序中一样的那些原子)
(4) 根据向量的排序,重新对原子进行排序
(5) 如果原子排序发生了变化,则从第 2 步开始循环
------- 循环一 ------------
(6) 首次到达这里,如果此时有原子排序一样(即已收敛),则认为他们是对称情况
(7) 如果排序最大值小于原子数量,则需要“断交”,然后再从第二步开始循环
------- 循环二 ------------
(8) 所有原子都已有唯一排序
对该算法进行一一说明:
初始标识符和 ECFP指纹一样,根据以下几个属性
- 原子连接数
- 非氢化学键数
- 原子序号
- 原子电荷正负
- 原子电荷绝对值
- 连接氢原子个数
从中也可以看出之前提到的,与 CIP 很大不同是优先先比较连接数,因此乙醚中的甲基因为连接数最少会排最前面。其次比较非氢化学键,即价键,然后再是原子序号。由于初始化后无需考虑周边原子环境而应该直接对原子进行排序,因此刚进入循环时会跳过第二步,其次由于也没有“上次排序”,因此我们先看第四步,即根据排序结果给每个原子一个“排序”,如乙醚,则排序是1 2 3 2 1,两遍碳只有一个连接数,排序最小(即最高),中间三个连接度一样则碳更小。此时显然原子顺序发生变换,开始循环。
如果了解 ECFP 则知道下一步就是将周边原子信息作为该原子信息,一般会采取加和、组合这样的操作。但这里用“质数积”这么一个操作以保证减少“撞车”(即原子环境不同但加在一起却相同),质数表大致是:[2, 3, 5, 7, 11, 13, 17, 19, 23 ...],所以原来原子排序转化为质数则是2 3 5 3 2,质数积则是3 10 9 10 3。
1 | 原排序 1 2 3 2 1 |
此时进入第三步需要排序,现在要注意那句话维持上一次的排序,虽然质数积中氧并不是最大值,但上次排序中氧是最大值,因此氧与碳的排序并不会变化。第四步重新对原子进行排序仍然是1 2 3 2 1。我们可以这样理解该算法啊,质数积的目的是让原来排序相等的原子之间进一步根据环境来较量谁更高,而原来就已分胜负的原子间不会再较量。因此第五步判断发现原子排序未发生变化,跳出循环一,进入第六步,因首次跳出循环一,认为排序相同的原子属于对称情况,然后进行断交。
断交过程就是让排序一样的原子出现一个大一个小,这样就可以继续循环二,最终可以保证所有原子都有排序。可能有人觉得既然排序一致说明是对称,而对称意味着无论取哪个原子都行,即排序无所谓,为什么还要断交?断交的目的是为了防止对称导致的“失去方向”。以文献中的“经典”例子说明:
立方烷是高度对称的一个结构,其8个碳的标识符完全一致,所以确实无论哪个碳作为“第一个碳”都没有任何问题,但是我们如果不断交,则没办法画出这个分子。确实,我们可以随便找一个碳作为第一个(大哥),可然后应该谁做第一个小弟呢?剩下的小弟可不是都和这个大哥关系好的,毕竟有人和大哥直接相连,有人不直接。因此我们让“大哥”排序小 1,而其他所有人的排序都增大为原来的两倍,则再次循环就知道谁和大哥亲了(毕竟离大哥直接相连的原子下一步“质数积”会相对小一点)。
当排序解决了,生成 SMILES 自然就很容易了,即先从排序最小的原子开始,如果它只有一个键,则下一个接上该原子(无视此原子排序),如果他有两个或以上的键,则选择原子排序最小的作为直接相邻的原子(当然,是需要括号的,因为产生了支链)。以丙酮为例:1
2
3 C - C ( = O ) - C
10106003 30406000 10208000 10106003
1 4 3 2
不难发现第一个碳排序最高,因此其为 0 号原子,由于第二个碳与之直接相连,则其为 1 号原子,该碳又连着氧和碳,根据排序碳靠前,则末尾的碳是 2 号原子,氧是 3 号原子。最终 SMILES 是CC(C)=O。
环的处理
在 CANGEN 原文给出基于深度优先的搜索策略,还是以立方烷为例,第一个原子是图中的C12(这里12指的是 SMILES 中的“闭环键”的标记,不是表示原子序号),C3、C25、C1都与C12直接相连,按照上述的排序规则,C3>C1>25按理说会想到用括号表示C12(C3..)(C1..)C25..,但所谓深度优先搜索,即我们会先从C3开始走下去,下一个是C4(里面的C3排序比C4低,所以先走C4)。最终顺序就成了图中的样子,且因为所有的原子都已走到,因此最终就变成了C12C3...C1...C25,无需再用括号。
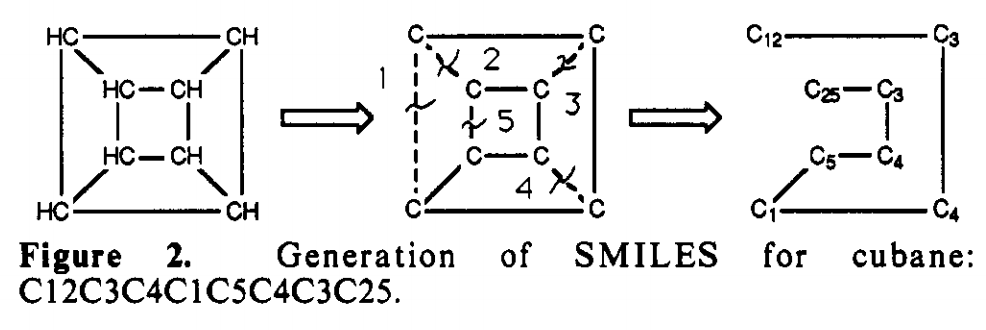
当然这个方法已经比较古老了,如果完全尊从该算法是无法考虑手性的,而且效率并不高。现在 RDKit 已经采用效率更高,且可能考虑手型构性甚至“相对手性”的结构,详情可以阅读该论文,其优点是对于已经明确排序的原子不会再计算其周边原子情况,浪费计算量。另外 O’Boyle 又借鉴 InChi 的标准化方法来处理 SMILES(详见论文),不过笔者尚未深入研究。