贝叶斯优化在化学中的应用-条件优化
Comment在一个很大搜索空间内想找到最优的参数,一般会有模拟退火、遗传算法等基于随机的算法,但这些算法收敛性较差,需要很大的迭代次数才能达到最优,对于迭代成本非常高的一些应用场景,这些方法恐怕不是很适用,化学反应中的条件优化就是这么个迭代成本非常高的场景。常规的一次化学实验就需要花费 4-12 小时才能得到比较可靠的收率数据,假设一天可迭代两次,迭代 10 次需要一周的时间。而由于没有大量数据直接构建收率与条件参数的数学模型,自然也无法使用梯度下降法之类的方式。贝叶斯优化十分适用于此类问题,通过小样本建模“推荐最有可能得到高产率的条件参数”。
对于一般的化学反应,需要优化的无非是温度、时间与浓度,这些参数一是相对独立,可以通过单因子分析得到一般规律,二是本身也确实存在一定规律,我们知道温度越大反应越剧烈,产生副产物的可能性越大,必然存在一个最优值;时间也是如此,越长则反应越充分,但有时可能随着时间推移产物慢慢会分解或者进一步发生副反应。如果是个特殊的催化反应,则往往兼容性较好,时间过长也无副产物,那么一般无需优化该变量。
往往最令人头痛的是一些金属催化反应,所需要使用的催化剂及配体范围很大,如最常见的 C-C 偶联反应 Suzuki-Miyaura 以及 C-N 偶联反应 Buchwald-Hartwig 常见配体可有几十种之多,难以捉摸规律,合成人员也很难甄别当一个反应收率不高或者压根没有反应到底是催化剂配体没有选对还是该底物确实很难合成,是应该继续尝试更多条件还是及时止损换一种合成方式,亦或者换其他条件参数,比如碱的强弱、当量浓度等。相比与温度与时间,催化体系与碱以及溶剂都可能存在相关性,导致无法使用常规的单因素法进行高效筛选。
接下来我们以2021年发表在《自然》的论文《Bayesian reaction optimization as a tool for chemical synthesis》研究贝叶斯优化在条件优化中是如何应用的[1]。
参数特征
贝叶斯优化中需要构建高斯过程模型作为”代理模型“以预测不同条件参数下收率的情况,为了描述一个化学反应所使用的条件,我们需要对条件参数特征化。论文中使用了以下方法将配体、碱、溶剂、浓度还有温度进行特征化。由于配体、碱以及溶剂都是化合物,则使用分子描述符以及量化特征。分子描述符一般是些预测的理化性质,如油水分配系数(logP)、拓扑极性表面积(tPSA)、原子频数信息,如 sp3 碳数量,以及一些预测的电子性质,已有比较久的历史,可使用一些开源的软件包进行计算,如 Python下可以用 Mordred,也能使用基于 java 的 Padel-Descriptor 等。量化特征一般是使用 xTB、Gaussian 等软件计算分子的电性、结构以及能量特征,比如偶极矩、分子体积、LUMO、HOMO 等。论文作者一共计算了 300+ 描述符,但似乎没有给出具体计算这些描述符的脚本。笔者了解计算量化描述符的化可以考虑使用 Multiwfn。
代理模型构建
作者使用 GPytorch 包实现高斯过程,从名字可看出这是个利用 Pytorch 进行 GPU 加速的高斯过程包。以下是一个简单的实现方法,初始化该模型之前需要先初一话一个 likelihood 实例,默认使用 GaussianLikelihood,用以计算 $ p(y|f(x)) $
1 | class ExactGPModel(gpytorch.models.ExactGP): |
训练模型时需要定义一个优化器,一般是 Adam;一个核函数,用的比较广泛的是径向基函数(RBF),但是本文的作者采用 Matérn52 kernel,该核是 RBF 的超集,其中的重要参数 nu = 2.5 (如果 nu = 0.5 则就是 RBF[2]),但是文中并没提具体原因;还有一个损失函数,一般是为了最大化边际似然(marginal log likelihood)。
训练过程和 Pytorch 相似,以下是 GPyTorch 的 官方示例。1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29import os
smoke_test = ('CI' in os.environ)
training_iter = 2 if smoke_test else 50
# Find optimal model hyperparameters
model.train()
likelihood.train()
# Use the adam optimizer
optimizer = torch.optim.Adam(model.parameters(), lr=0.1) # Includes GaussianLikelihood parameters
# "Loss" for GPs - the marginal log likelihood
mll = gpytorch.mlls.ExactMarginalLogLikelihood(likelihood, model)
for i in range(training_iter):
# Zero gradients from previous iteration
optimizer.zero_grad()
# Output from model
output = model(train_x)
# Calc loss and backprop gradients
loss = -mll(output, train_y)
loss.backward()
print('Iter %d/%d - Loss: %.3f lengthscale: %.3f noise: %.3f' % (
i + 1, training_iter, loss.item(),
model.covar_module.base_kernel.lengthscale.item(),
model.likelihood.noise.item()
))
optimizer.step()
贝叶斯优化与实验设计
上篇博客 提过,所谓贝叶斯优化,则是利用代理函数(高斯过程)的预测结果,通过采集函数得到下一个推荐样本,在化学反应条件优化的应用中则是选择下一个待测实验条件。采集函数一般有 PI 和 EI 两种算法,PI 是得到采样点比已知最大点还要大的概率最大,EI 则是希望采样点比最大点还要大的部分的数学期望最大。作者在文中比较了 EI、汤普森采样、贪心算法、以及均值最大(Exploit)、方差最大(Explore)几种采样方法,发现 EI 的方法是最好的。
另外,由于传统的贝叶斯优化法只能得到一个推荐参数,作者在这里做了一个调整,使用 Kriging Believer Algorithm,从 源码 看大概意思是先通过采集函数选择一个样本(条件),然后将该样本(包括其预测结果)放到训练集中重新进行 GP 模型构建以得到下一个样本,如此迭代直到得到所有样本。该方法应该等同于 Efficient Global Optimization。
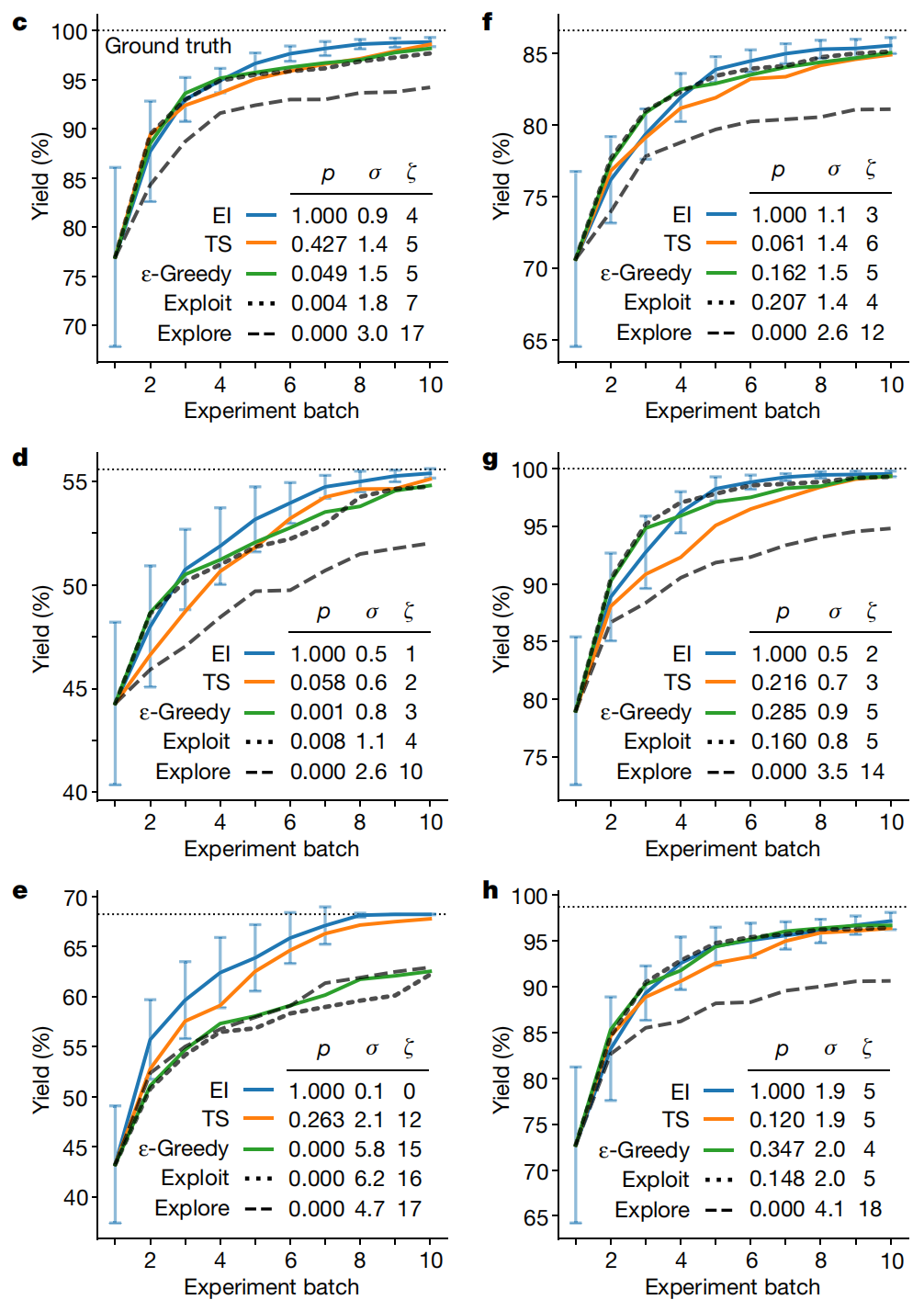
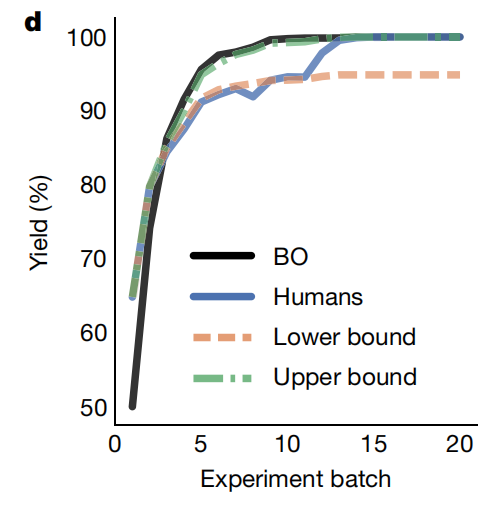
有意思的是最后作者还做一个比赛,让化学家们和BO进行比较,结果发现最开始的时候化学家的收率比较高,可能因为人有先验知识,但是BO能以可快的速度找到最优解,最终还是要比人厉害一些。
尽管看上去很棒,笔者还是有一些地方对该文有些疑惑甚至质疑,笔者了解该方法不能冷启动,而需有一些 Ground truth 作为早期的训练集,那意味着优劣参半的一些因素:
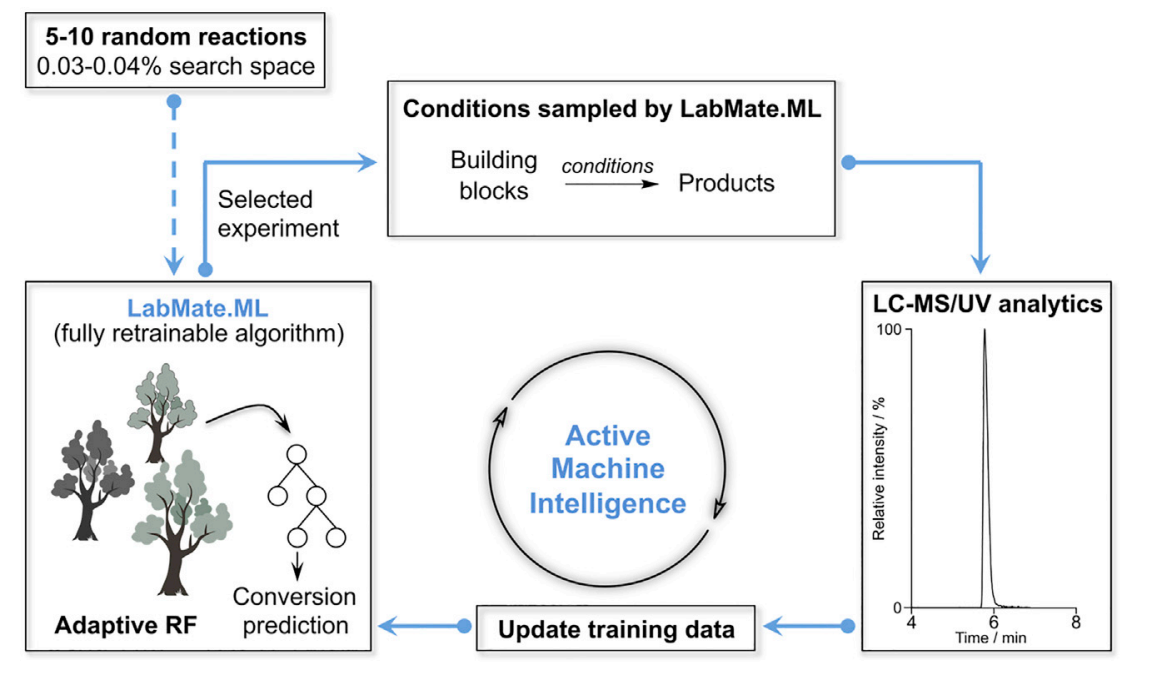
如果测试集的底物与训练集相近,那么可能建模效果会比较好,第一次实验可能就能获得比较好的结果;相反如果测试集和训练集不相似,可能甚至会有反作用,即使经过几次实验迭代仍然因为被带偏而结果不好。文中可能都选择一些较为简单的化合物,所以整体上没有这种问题,从结果可看出每次迭代收率都是在进步的,但真实应用中是否需要根据测试集的分布选择更好的训练集,还是如其他机器学习一样,数据是越多越好。说到冷启动,之前有论文[3]就有提过,只需要5-10个随机的实验,即可建模进行条件优化,该模型虽未用到贝叶斯优化,但也有许多相似之处,用的是随机森林回归作为代理模型,用 Exploit 方法进行采样,同样得到很好效果,(不过改论文优化的参数比较粗糙,看作者背景似乎不研究化学…)
写在最后,真实的情况往往比这个更加复杂,对于早期未知的化学反应,我们无法知道这个反应到底能否进行,并非所有反应都能达到很高的收率;收率不高不一定是试剂选择不对,可能有其他因素,包括试剂的性状质量(有时不得不买进口试剂)、检测原因,比如有些物质没有紫外吸收,用常规 LCMS 方法无法观测到产物;有些物质容易分解;反应体系的水氧含量过高等等,这些原因会导致实验失败。在其他因素已经排除,仅针对一些空间很大的条件(如催化剂、碱、温度等)进行优化时效果会比较好,未来配合上高通量的自动化反应,想必条件优化会更为边界。当然收率的优化可能主要用在化学工艺研究,那便还需要面临放大效应、局部效应等因素,使得条件优化过程变得相当复杂,比如克级下的最优条件不一定在于公斤级最优,通过 BO + 公斤级实验来回折腾可能不如有经验的工艺化学家通过现象观察原因最终进行调节来的有效。
参考资料
[1] https://doi.org/10.1038/s41586-021-03213-y
[2] https://www.guyuehome.com/34071
[3] https://doi.org/10.1016/j.xcrp.2020.100247