信息增益与主成分分析在化学信息学中的应用
Comment摘要:信息增益一般用在数据挖掘中的决策树方法领域,它是指当前熵与两个新群组经加权平均后的熵之间的差值。
在化学信息学中,我们可以利用信息增益判断分子指纹的每个子结构对分子活性的影响程度。
主成分分析是数理统计中的一种技术,意在降低变量的维度,防止过拟合现象以提高模型的准确度。
在化学信息学中,我们可以利用主成分分析技术解决分子描述符太多且可能存在相关性的问题。
本文简单介绍上述两种方法的原理,并描述他们在化学信息学中的具体应用。
1. 化学信息学与分子描述符
化学信息学(chemoinformatics)一词最早由Brown在1988年提出,“化学信息学实际上是一种信息源的混合体。它可将数据转换为信息,再由信息转换为知识,从而使我们在药物先导化合物的识别和组织过程的决策变得更有效。”[1]化学信息学将统计学、数据挖掘的方法应用在化学数据上,建立数学模型以预测一些化学性质。尽管这个词1988年才被提出,早在上世纪60年代,就有人开始定量地研究化学结构和他们活性之间的关系,最有名的便是1964年Hansch和Fujita以及Free和Wilson发表的关与分子描述符的研究[2,3],他们的研究为之后的定量构效关系(QSAR)研究打下了基础。之后,越来越多的人投入了QSAR的研究,描述符也从原先最基础的理化性质,扩展到拓扑性质、立场性质等等,现在已经可以计算数以千计的分子描述符。
所谓分子描述符,就是描述分子的一种方法,就如我们可以通过“身高、体重、年龄”来描述一个人一样,我们也能通过一些描述符来描述一个化合物。那么,每个分子就可以认为是化学空间中的一个点,这个点的坐标就是这个分子的各个描述符。这样,我们就可以把所有分子都通过一定的描述,映射到一个化学空间中,这个空间包含了所有我们知道的信息。
上述的分子描述符一般为连续的实数(如脂水分配系数等)或者是自然数(电荷数、环的数量等),虽然具有一定的物理意义,但是由于描述符数量多,挑选出“优秀”的描述符非常困难。除了上述方法,还有一种描述分子的方法是指纹描述符,即将每个分子都描述为一个一定长度的二进制片段,这个二进制片段可以理解为一个指纹字典,每个“位”都指代一种特征,如果分子符合该特征则表示为1,反之为0。由于每个分子之间的结构与拓扑学性质不同,他们的指纹也不相同,所以可以作为一种描述分子的方法。
2. 信息增益算法及其在化学信息学中的应用
信息增益是指当前熵与两个新群组经加权平均后的熵之间的差值,他可以用来评估化合物分子的子结构对于活性的影响强弱,进而减少不重要的子结构信息对于预测结果的影响。比如当我们发现无论化合物中是否有酚羟基,活性都有高有低均匀分布,那么它就是不重要的子结构信息。通过信息增益的计算,我们便能区分哪些子结构信息是重要的哪些是不重要的。
2.1 信息增益介绍
信息增益,所增益的是信息的复杂程度,我们一般用信息熵来表示,公式如下:
$$ Entropy(s) = -p1 * Log2(p1) - p2 * Log2(p2)$$
这里的p1,p2指的是阳/阴性率。比如,如果100个化合物中,有50个是阳性的,50个是阴性的。那么这个体系的总熵就可以将p1 = 0.5 , p2 = 0.5带入上述公式,得到Entropy(s) = 1。接着计算某个子结构T的信息增益值,方法是计算出所有含有羟基的分子的信息熵(用Entropy(t)表示)以及不含有羟基的分子的信息熵(用Entropy(t’)表示)。与上述步骤一样,我们同样可以统计出含有羟基的分子有多少是阳性的,有多少是阴性的进而算出Entropy(t)。不含羟基的分子的熵也能计算出。将这两个情况的熵加权求和就能得到“羟基的信息熵”,加的权则是含羟基的分子在所有分子中所占的比例Pt。用公式表示如下:
$$ Entropy(T) = Pt * Entropy(t) + (Pt’) * entropy(t’) $$
将总的信息熵减去羟基的信息熵,则得到羟基的信息增益值
$$ IG(T) = Entropy(s) - Entropy(T) $$
信息熵的概念源于信息论,借用热力学中熵的概念,这里是指信息的复杂程度,从公式1中可以看出,如果p1=p2=0.5,即阳性与阴性的概率各占一半,完全是随机的,所以此时信息熵最大为1。如果p1或p2为0,那么说明该体系下全是阳性或阴性,此时信息熵最小为0。Entropy(s)可以理解为原始数据的信息熵,Entropy(T)则是通过判断是否含有T基团将原始数据拆分为两个数据集的熵的加权和。如果拆分后熵变的很小,信息增益值就会很大,说明这个基团对于活性的决定性很大。若Entropy(T)=0,即t及t’的熵都为0,说明一个全都是阳性集,一个全是阴性集,换言之通过判断是否有T基团成功地分离出活性数据集和非活性数据集,说明T对活性起到决定性因素。
2.2 信息增益在化学信息学中的应用
信息增益在化学信息学中的应用主要包括分子指纹字典(子结构)的评估,决策树分类模型的构建。
子结构评估
对于数据量比较少的数据集,并非分子指纹字典中所有的子结构都需要用到,很多子结构对于活性几乎没有影响,于是我们需要过预先过滤这些子结构[4]。如果不删掉可能对于后期的模式识别效果会有影响。所以在建立分类模型之前,往往会先通过设定信息增益阈值,去掉一些不重要的子结构信息。当然,阈值的确定可以通过对模型的交叉验证得到。当我们进行毒性预测的时候,这些“影响力”大的结构可以认为是需要警惕的结构,这样有利于进一步降低毒性的优化。
决策树分类模型构建
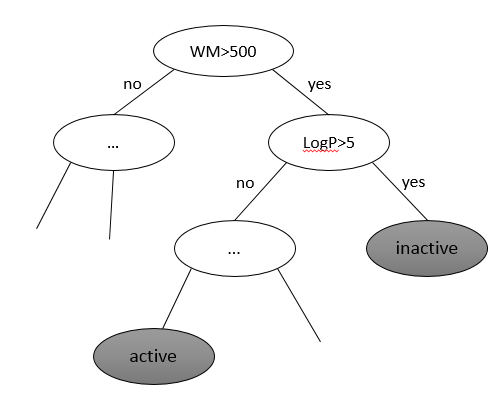
决策树是一种容易理解却广泛使用的分类方法,属于机器学习范畴。机器通过考察训练集分子的描述符性质,构建一棵决策树分类模型。模型内容就是一棵树的形状,每个节点即一个描述符的判别式,从根节点出发,最终落入一个叶节点为分类的结果。如这样一棵树,输入一个分子,首先询问分子量是否大于500,如果大于500则再问LogP是否大于5,如果仍然为是那么输出结果“无活性”,如果小于5则继续询问。(见图2-1)构建这样的树最大的问题莫过于如何选择描述符的判别式,为什么是分子量大于500不是大于400,为什么先问分子量再问LogP?决策树分类模型计算不同判别式的信息增益值,增益越大说明该信息越重要,就越要先问。所以在上述例子中,一定存在IG(M>500) > IG(LogP>5);IG(M>500) > IG(M>400)等等。需要注意的是,从2.1章可以知道,IG(T)就是IG(分子存在T结构) = IG(分子不存在T结构),同样IG(WM>500)和IG(WM<=500)也是一样的。所以信息增益的自变量在化学信息学中可以是描述一个子结构是否存在,也可以描述某个分子描述符是否满足某个判别式,互补的两个判别式没有区别。
3. 主成分分析方法及其在化学信息学中的应用
主成分分析,通过数学手段将原来的许多中可能有内在线性关系的变量降维到较小的变量,这样可以避免因为变量之间有联系导致回归结果中放大了某些变量的重要性,因此可以通过这种方法提高模型的预测能力。在化学信息学中,我们常能通过许多不同的描述符来描述一个分子,目前可以计算出的描述符估计有2000多个,而这些描述符之间可能会存在一定的关联,除了通过手动挑选描述符以减少变量外,主成分分析法可以方便有效地避免变量关联导致的过拟合现象。
3.1 主成分分析算法简介
首先对下标进行解释,假设共有n个化合物,m个描述符。i代表第i个化合物,j代表第j个描述符。每个化合物的每个描述符的值用x(i,j)表示,即得到一个n*m的变量矩阵X
主成分的计算过程[5]分为以下5个步骤:
1)对原始数据进行标准化即$x ̃(i,j) = [ x(i,j) - x*(j) ] / s(j)$,其中x*(j)是第j列变量的平均数。这样X ̃中每列的变量都满足平均数为0方差为1。这样的好处是所有描述符的方差都一样,进一步计算协方差阵、相关阵会变得非常容易,且计算本身并没有丢失变量本身的价值。
2)计算相关矩阵即$R = X ̃^T X/(n-1)(R(j,j’) = Σi[x ̃(i,j) * x ̃(i,j’)] / (n-1))$,相关阵可以理解 R(j,j’)即第j个描述符与第j’个描述符的皮尔逊相关系数。
3)对R进行特征分解 R = UAUT,其中A = diag(λ1,λ2…),λ 即为 R 的特征值。求解时要求 λ1>λ2>… 。U是每个描述符的特征向量,第j列即第j个描述符的特征向量。
4)用特征向量描述原变量,即 ξ(j) = U(j)Y,这里的Y即主成分。可以把主成分想象成一个新的描述符组,那么U就是让主成分映射到原描述符的变化矩阵。上式还可以简化为 ξ = UY。 由于U是正交阵,所以可以得到Y = Uξ,即可以从原描述符组ξ映射到主成分Y。
5)计算 (λ1+λ2+λ3+…+λj) / Σλ 得到前j个主成分的贡献率来判断要选取几个主成分。假设原来有10描述符即可以得到10个主成分,但一般我们可能会发现前5个主成分已经有80%的贡献率,那么我们可以选择前5个主成分作为描述符变量,这样成功的缩小一般的变量数,而“信息量”并没有损失太多。
3.2 主成分原理及其几何意义简介
图3-1是两个变量(x1,x2)的分布情况,可以看出如果直接删去某个变量,信息都会损失非常多,因为数据在两个方向上的分布都比较分散。如果能建立一个新的正交坐标体系,使得数据在y1上的离散程度最大(方差最大),在y2上的离散程度则较小,那么我们便(认为)可以考虑仅用y1表示这些数据可以成功的对数据进行降维处理。(那么我们可以仅用y1 来表示这些数据,这样可以成功的对数据进行降维度的处理)。方法是旋转这个坐标轴。
y1 = x1 cos(θ) + x2 sin(θ)
y2 = -x1 sin(θ) + x2cos(θ)
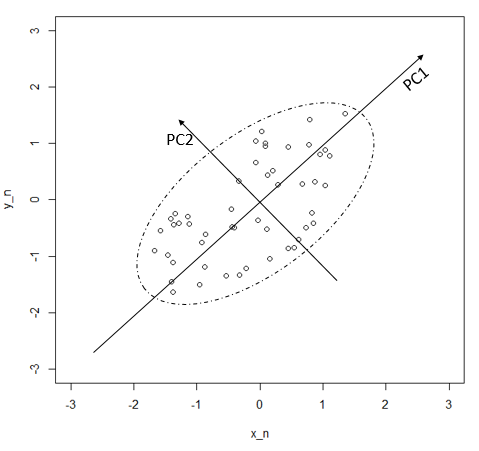
可以看出,坐标经过旋转以后,原数据在y1上的离散程度很大,即使没有了y2,也能很好的描述这些数据的特征。从这个简单的例子可以看出,主成分分析的过程就是坐标系旋转的过程[6],通过对坐标系的旋转,使得在保持坐标系之间保持正交的基础上,可以让数据在这些坐标上的离散度有最大、次大等等之分,这样我们便可以忽略“不重要”的维度。举个现实生活中的例子,我们看到的世界是三维,如果把地面北面作为x轴,东面作为y轴,垂直方向作为z轴,那么每个物体都有其坐标。而然当我们想要用照相机拍摄这些物体的时候我们不得只能用2维的图片描述这个三维的世界,那么我们无论是从前视角(忽略y轴),侧视角(忽略x轴)还是俯视角(忽略z轴)都不能很好的拍出真实的情况。可聪明的摄像师会选择在高空斜视的方式尽可能把每个物体的特征都表达出来,那么它选择的这个角度就是他认为的“主成分”。
主成分也有其缺点,那么就是这些主成分本身没有意义。就如上述例子,如果我们把y2的成分去掉仅保留y1,那么这条线即不是x轴也不是y轴,这条线上的点也不再是原来原始数据上的点。同样的,照片里的坐标点也是没有意义的,由于降维导致的“失真”,使得你无法通过照片里的像素回溯到真实世界里面的具体坐标。在化学信息学中其缺点更显而易见,原来不同描述符有其固有的物理、化学、拓扑学等含义,而通过纯粹的数学手段将他们视为坐标系进行“旋转”,自然不再有任何现实意义。
3.3 主成分分析在化学信息学中的应用
主成分分析在化学信息学中的应用非常多,作为一种数学统计方法,它可以方便地将许多变量缩小到几个变量,不仅能提高预测模型的准确率,也能方便研究者进行进一步分析。在化学信息统计分析方法上,通过PCA可以将变量降至二维,这样便可以直观地从图中对数据进行分析。John R. Owen 等人运用PCA技术将描述符、分子指纹进行降维,并可视化分析数据的聚类分布情况。[7]在具体的应用中,比如观察排放化合物对环境的影响。PCA可以把化合物成分作为变量,将大量化合物降维成几个主要的“化合物成分”,这样可以分析哪些化合物对环境的影响起到关键的作用,有利于进一步对环境优化政策进行指导。[8]在药物设计领域,PCA还可以用在动力学模拟的分析,将分子的构象[9]甚至是原子的波动情况[10]进行主成分分析。
4. 总结
在化学信息学中,有许多我们可以用到的数学、计算机科学的方法,有些可以用来建立模型(如机器学习方法,回归分析等)、有些可以用来便于我们统计(PCA,显著性水平计算,置信度等),也有些用来提高一些统计、运算、挖掘速度的算法(遗传算法、神经网络算法等)。了解这些方法对化学信息学研究有着非常重要的意义。本文简单介绍了信息增益方法与主成分分析方法的计算过程及其原理,并且讲述一些他们在化学信息学中的具体应用案例。
参考文献
[1] Brown F K. . Chemoinformatics: What is it and How does it Impact Drug Discovery[J]. Annual reports in medicinal chemistry, 1998, 33: 375-384.
[2] Hansch C, Fujita T. p-σ-π Analysis. A method for the correlation of biological activity and chemical structure[J]. Journal of the American Chemical Society, 1964, 86(8): 1616-1626.
[3] Free S M, Wilson J W. A mathematical contribution to structure-activity studies[J]. Journal of Medicinal Chemistry, 1964, 7(4): 395-399.
[4] Shen J, Cheng F, Xu Y, et al. Estimation of ADME properties with substructure pattern recognition[J]. Journal of chemical information and modeling, 2010, 50(6): 1034-1041.
[5] 刘建平, 朱坤平, 陆元鸿. 应用数理统计[M]. 华东理工大学出版社. ISBN: 978-7-5628-3264-5/O·244
[6] 汪冬化. 多元统计分析与SPSS应用[M]. 华东理工大学出版社. ISBN: 978-7-5628-2874-7/F·228
[7] Owen J R, Nabney I T, Medina-Franco J L, et al. Visualization of molecular fingerprints[J]. Journal of chemical information and modeling, 2011, 51(7): 1552-1563.
[8] Kong L, Kadokami K, Wang S, et al. Monitoring of 1300 organic micro-pollutants in surface waters from Tianjin, North China[J]. Chemosphere, 2014.
[9] Xiao X, Zeng X, Yuan Y, et al. Understanding the conformation transition in the activation pathway of [small beta]2 adrenergic receptor via a targeted molecular dynamics simulation [J]. Physical Chemistry Chemical Physics, 2015,
[10] Chen J, Liang Z, Wang W, et al. Revealing Origin of Decrease in Potency of Darunavir and Amprenavir against HIV-2 relative to HIV-1 Protease by Molecular Dynamics Simulations [J]. Sci Rep, 2014, 4